- Home
- News
- Spotlight on Science
- X-linked intellectual...
X-linked intellectual disability related to a mutation in the catalytic domain of O-GlcNAc transferase
16-08-2019
O-linked beta-N-acetyl-D-glucosamine (O-GlcNAc) transferase (OGT) mediates protein O-GlcNAcylation, a regulatory modification abundant in the brain. Recently, OGT mutations were linked to intellectual disability. A mutation of N567K OGT has now been shown to disrupt its acceptor binding site, revealing the mechanisms underlying pathogenesis.
O-linked β-N-acetyl-D-glucosamine (O-GlcNAc) transferase (OGT) and O-GlcNAcase (OGA) mediate an essential post-translational modification of cytosolic proteins (O-GlcNAcylation), regulating different aspects of cell physiology ranging from cell cycle and transcription to protein trafficking and stability. To date, thousands of O-GlcNAcylated proteins have been identified in the mammalian brain, and O-GlcNAc has been shown to have a crucial role in synaptic activity and plasticity, learning and memory formation. In addition, perturbations in protein O-GlcNAcylation have been linked to neurodegenerative disease and, more recently, to neurodevelopment.
Intellectual disability is a spectrum disorder defined by early-onset impairment of cognitive function and limitation of adaptive behaviour. The most common causes of intellectual disability include monogenic mutations in more than 650 genes. Recently, mutations in OGT have been linked to intellectual disability. Previously reported pathogenic OGT mutations had marginal effect on OGT activity and were speculated to influence OGT protein-protein interactions. However, little is known about the underpinning mechanism. In concomitance with mouse embryonic stem cells and Drosophila melanogaster studies, structural biology was used to identify the molecular basis of the first intellectual disability-associated mutation in OGT catalytic domain (c. X:70779215 T > A, p. N567K) implicating O-GlcNAcylation in neurodevelopment.
OGT comprises two parts: an amino-terminal regulatory part encompassing 11.5 tetratricopeptide repeats (TPRs), which form a 120-Å superhelix and serve as a potential interaction surface for substrates and binding partners, and a bi-lobal Rossman fold at the carboxy-terminus, which is interspaced by a domain of unknown function, that is evolved to catalyse O-GlcNAc transfer from UDP-GlcNAc onto serine or threonine residues of acceptor proteins.
The mutated residue N567 resides in a highly conserved loop in the OGT catalytic domain, borders the −2 subsite as part of the OGT acceptor substrate binding cleft. The N567K mutation was hypothesised to cause a protrusion in the substrate binding pocket, thereby altering the substrate binding ability of the N567K OGT. Indeed, in vitro analyses showed reduced catalytic activity of the mutated enzyme.
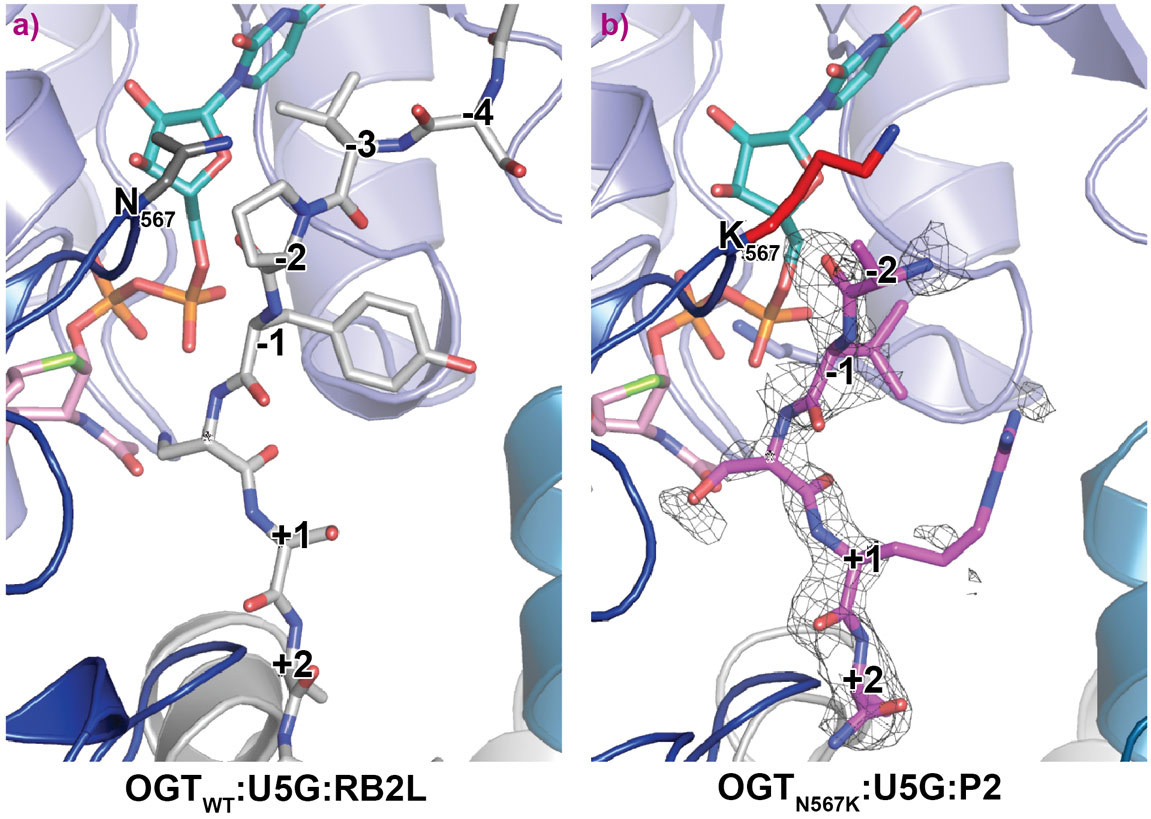
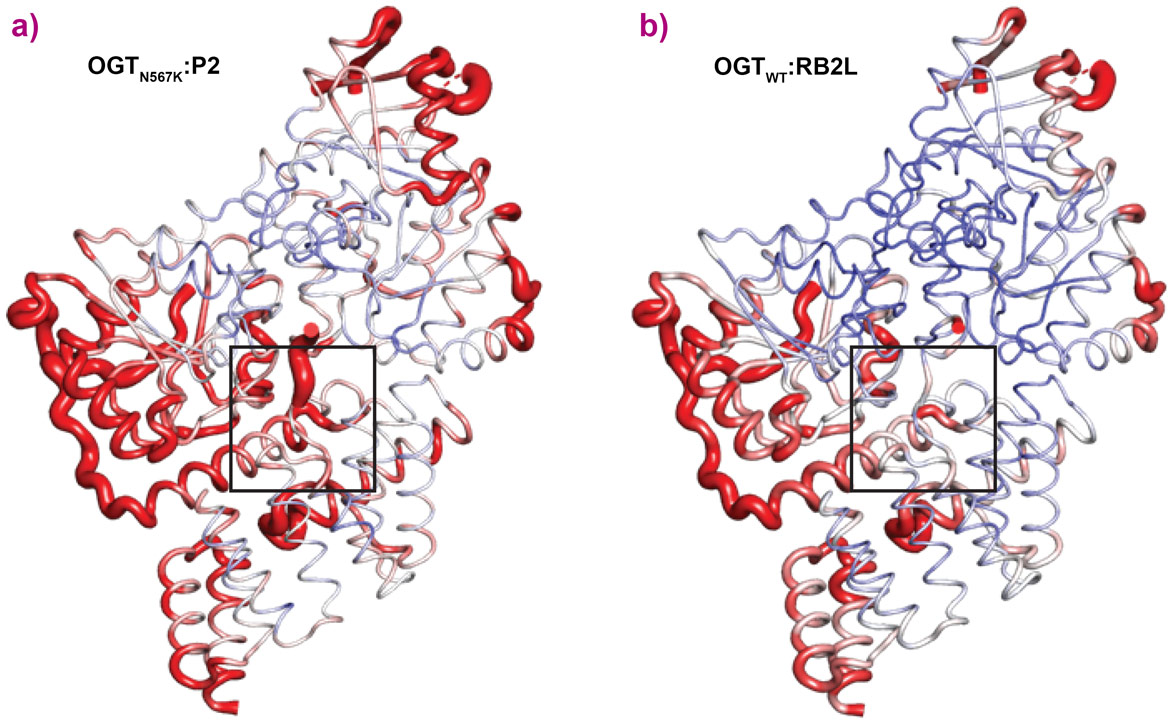
OGTN567K was then co-crystallised with a donor analogue, UDP-5S-GlcNAc, and two OGT acceptor peptide substrates, P1 (KKVPVSRA) and P2 (KKVAVSRA). Only the condition containing P2, which contains an alanine at the −2 subsite, yielded crystals. The X-ray diffraction experiments performed at beamline ID23-2 allowed structure solution by molecular replacement. The OGTN567K structure superposed onto OGTWT (PDB ID code: 5C1D [1]) with an RMSD of 0.4 Å across 675 Cα atoms, suggesting the mutation does not induce large conformational changes in the catalytic domain. Initial unbiased difference electron density maps revealed partial density corresponding to the acceptor peptide (Figure 1). Remarkably, this only covered the P2 backbone in the +2 through to −2 subsites, suggesting that the N567K mutation may induce increased flexibility at the N-terminal tail of the acceptor peptide (Figure 1). In agreement with this, a comparative B-factor analysis suggested that the acceptor peptide bound to OGTN567K is substantially more flexible than that bound to OGTWT (Figure 2). Close inspection of the OGTN567K acceptor substrate binding cleft revealed that the protrusion caused by the N567K mutation partially occupies the position of the −2 proline frequently observed in OGT acceptor peptides. In summary, the N567K mutation appears to disrupt the OGT acceptor binding site.
 |
|
Figure 1. Crystal structures of OGT ternary complexes of the OGTWT and OGTN567K active site in complex with UDP-5S-GlcNAc (U5G, turquoise, orange atoms) and an acceptor peptide. a) OGTWT is shown in complex with RB2L (retinoblastoma-like protein 2 peptide, light grey carbon atoms) [PDB ID code: 5C1D (57)]; b) OGTN567K is shown with P2 peptide (pink carbon atoms) (PDB ID code 6IBO). K567 is shown in red. An unbiased Fo–Fc difference map before inclusion of the peptide is shown as a mesh. |
This structural finding was further used to understand the phenotypes seen in drosophila and mouse embryonic stem cell (mESC) models. In Drosophila melanogaster the potential disruption in the OGT acceptor binding site of the N567K mutation led to hypo GlcNAcylation, while in mESC the mutation was compensated by reduced levels of OGA. However, this compensatory mechanism between OGT and OGA was not sufficient to avoid defects in neural differentiation, since the N567K mESC cells showed reduced neurite outgrowth compared to wild type cells. Thus, the N567K mutation may affect the O-GlcNAc proteome globally yet leads to a similar clinical phenotype, suggesting it is the catalytic activity/dosage of OGT that is important.
 |
|
Figure 2. Structural analysis of OGTN567K versus OGTWT ternary complexes. a) OGTN567K:P2 complex (PDB: 6IBO) shown in putty representation with the loop colour and radius both highlighted as a function of B-factors, with red colour and large loop radius indicating high B-factors (i.e. high positional uncertainty in the model). The acceptor peptide P2 is highlighted by the black square. b) Same as in (a), but showing OGTWT:RB2L complex (PDB: 5C1D). The acceptor peptide RB2L is highlighted by the black square. RB2L: Retinoblastoma-like protein 2. |
Principal publication and authors
Catalytic deficiency of O-GlcNAc transferase leads to X-linked intellectual disability, V.M. Pravata (a), V. Muha (a), M. Gundogdu (a), A.T. Ferenbach (a), P.S. Kakade (b), V. Vandadi (a), A.C. Wilmes (a), V.S. Borodkin (a), S. Joss (c), M.P. Stavridis (b), and D.M.F. van Aalten (a), Proceedings of the National Academy of Sciences 116, 14961-14970 (2019); DOI: 10.1073/pnas.1900065116.
(a) Division of Gene Regulation and Expression, School of Life Sciences, University of Dundee (UK)
(b) Division of Cell and Developmental Biology, School of Life Sciences, University of Dundee (UK)
(c) West of Scotland Genetic Service, Queen Elizabeth University Hospital, Glasgow (UK)
References
[1] S. Pathak et al., The active site of O-GlcNAc transferase imposes constraints on substrate sequence, Nat. Struct. Mol. Biol. 22, 744–749 (2015).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.